If We Didn''t Know How Brain Cells Were Dying, What Were We Actually Treating for 40 Years?
Summary
A previously undescribed neuronal death mechanism called "karyoptosis" was identified in Alzheimer''s disease and frontotemporal dementia patients by researchers at King''s College London, published in Nature Communications on June 25, 2026 — a discovery that challenges the foundational assumptions of four decades of dementia treatment strategy. Karyoptosis signatures were observed in 35% of frontal cortex neurons from Alzheimer''s patients compared to 15% in healthy elderly controls, confirming a statistically meaningful difference and establishing this mechanism as entirely distinct from apoptosis and necrosis, the two cell death pathways that had historically dominated scientific understanding of neuronal loss. This discovery provides a new explanatory lens for why anti-amyloid therapies — which absorbed $42.5 billion in private R&D over 25 years — achieved amyloid clearance but consistently failed to produce clinically meaningful cognitive improvement, a pattern confirmed by the 2026 Cochrane Review of 17 randomized controlled trials involving 20,342 patients. The appearance of karyoptosis in both Alzheimer''s disease and frontotemporal dementia raises a deeper question: whether these diagnoses share a common pathway of neuronal destruction that has gone entirely unrecognized for decades, and whether "Alzheimer''s disease" as a single diagnostic category is actually an umbrella term concealing multiple distinct pathological entities. With the p38 MAP kinase and LaminB1 protein interaction identified as a concrete molecular target — and a global dementia population projected to reach 152.8 million by 2050, generating a cumulative $14.5 trillion economic burden — this mechanism discovery may represent the beginning of a necessary paradigm shift in neurodegeneration research.
Key Points
Discovery of a Previously Unknown Neuronal Death Pathway
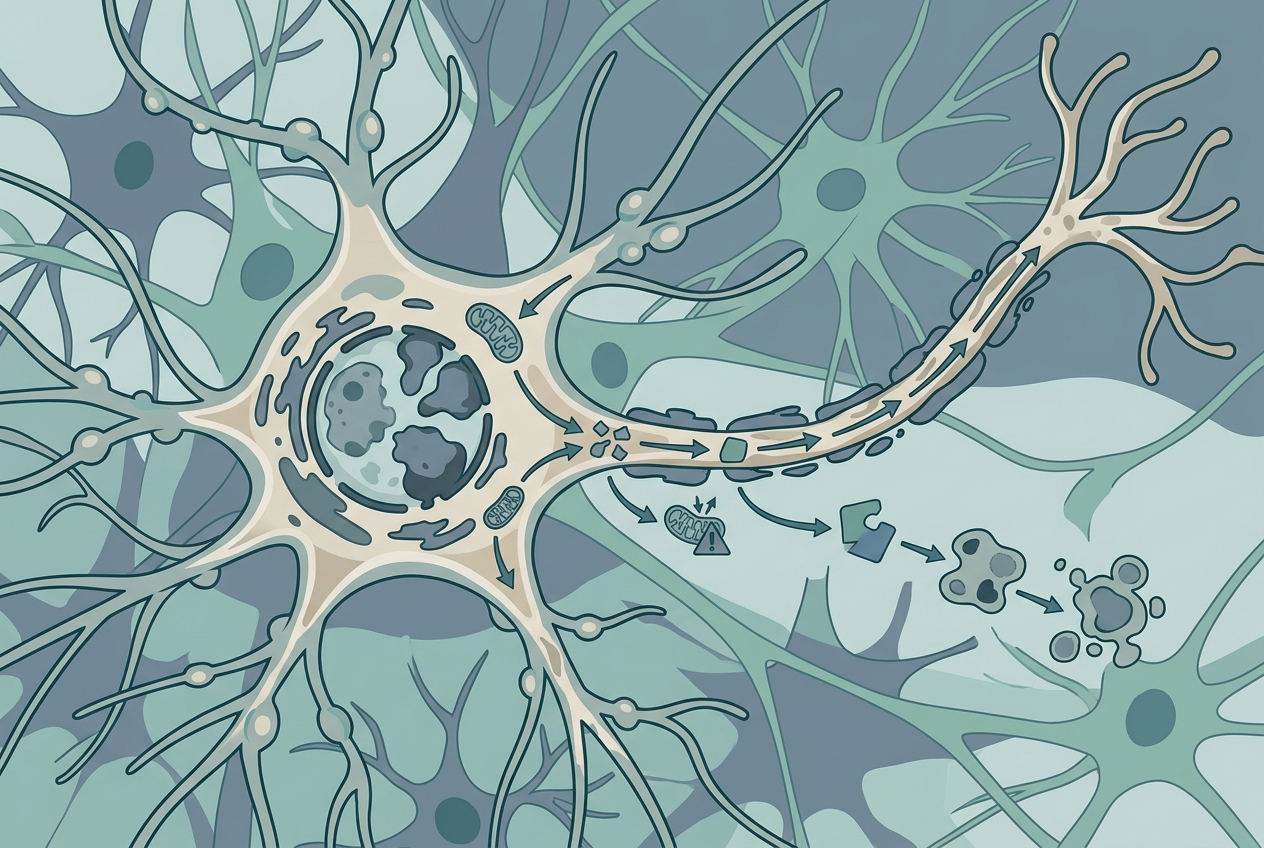
Dr. Manolis Fanto''s team at King''s College London capped a decade of research by identifying "karyoptosis" — a form of cell death in which nuclear envelopes shrink and then fragment under proteotoxic stress — a mechanism entirely distinct from apoptosis and necrosis, the two cell death pathways that had historically dominated scientific understanding of how neurons die. Karyoptosis signatures were found in 35% of frontal cortex neurons from Alzheimer''s patients, compared to 15% in healthy elderly controls, establishing a statistically meaningful elevation that cannot be dismissed as background noise or a minor incidental phenomenon. The research team analyzed roughly 3,000 cells from 28 patients using computational imaging algorithms and crucially confirmed the same mechanism in frontotemporal dementia, suggesting karyoptosis may represent a common neuronal death pathway across at least two distinct neurological conditions. Dr. Fanto described the finding plainly: "There are multiple ways in which neurons can die, and karyoptosis is one that was completely unknown before." This discovery was published in Nature Communications on June 25, 2026, with funding from the UK Dementia Research Institute and Alzheimer''s Research UK, and immediately attracted academic attention internationally. The implications extend beyond Alzheimer''s disease — if karyoptosis operates across multiple neurodegenerative conditions, similar undiscovered death pathways may be operating in Parkinson''s disease, ALS, and other conditions where unexplained neuronal loss is a defining feature.
40 Years of Amyloid Hypothesis — The Structural Failure Laid Bare
The amyloid hypothesis emerged in the 1980s as the dominant explanation for Alzheimer''s disease: amyloid beta protein plaques accumulate in the brain, trigger a cascade of neural damage, and produce the cognitive decline that defines the condition. This framework was compelling enough to attract $42.5 billion in private R&D spending over 25 years, generating a pipeline of anti-amyloid antibodies including aducanumab, bapineuzumab, solanezumab, lecanemab, and donanemab. The critical flaw was not that amyloid was irrelevant — these drugs successfully cleared amyloid from the brain — but that clearing amyloid did not reliably translate to clinical improvement for patients. Between 2002 and 2012, the clinical trial failure rate for Alzheimer''s drugs reached 99.6%, and the 2026 Cochrane Review of 17 randomized controlled trials involving 20,342 participants concluded that successful amyloid clearance "does not appear to be associated with a clinically meaningful effect," with a CDR-SB scale difference of merely −0.08, placing active treatment statistically indistinguishable from placebo. Karyoptosis provides an explanatory mechanism for this long-standing paradox: if a significant proportion of neuronal death occurs through an entirely separate pathway that amyloid-clearing therapy does not address, the absence of clinical benefit becomes logically predictable rather than merely disappointing. The Cochrane Review itself concluded that future Alzheimer''s disease-modifying research should focus on different mechanisms of action — a direction that the karyoptosis discovery directly answers.
p38 MAPK-LaminB1: A Concrete Therapeutic Target Emerges
The most immediately actionable finding from the karyoptosis study is the identification of a specific molecular mechanism amenable to pharmacological intervention: the interaction between p38 MAP kinase and LaminB1 protein, which appears to drive the nuclear fragmentation that kills cells through karyoptosis. In mouse neuron experiments, blocking this interaction prevented nuclear division and measurably reduced karyoptosis signatures — providing a manipulable causal handle on the mechanism rather than merely an observational correlation. Dr. Fanto noted that "specifically targeting the interaction of p38 MAP kinase with LaminB1 could slow the process of cell death, buying time for a more targeted treatment," framing the intervention potential in appropriately cautious but clinically meaningful language. What makes this finding immediately significant for the treatment pipeline is that p38 MAPK inhibitors already exist with documented human safety data: Neflamapimod is currently in Phase 2 for Lewy body dementia across 160 patients at 29 centers; MW150 ran Phase 2 data in Alzheimer''s disease; VX-745 has Phase 2 experience in mild cognitive impairment. Repurposing drugs with established human safety profiles toward a new mechanism eliminates the most time-consuming development phase — Phase 1 safety trials — potentially shaving years off the pathway to clinical testing. The fact that 33% of the 2025 Alzheimer''s pipeline already consists of repurposed agents confirms this is a well-validated strategy rather than a shortcut.
Alzheimer''s May Not Be a Single Disease — A Paradigm Challenge
The observation that karyoptosis appears in both Alzheimer''s disease and frontotemporal dementia — two conditions classified as pathologically distinct entities — raises what may be the most consequential intellectual challenge this discovery presents: whether "Alzheimer''s disease" as a diagnostic category actually corresponds to a single coherent biological disease. FTD affects 10 to 20% of all dementia patients worldwide, with an incidence of 2.28 per 100,000 person-years and a prevalence of 9.17 per 100,000, and is the second most common cause of dementia in people under 65. Pathologically, FTD involves different protein aggregates — TDP-43 or tau — and affects different brain regions than Alzheimer''s, which makes the shared karyoptosis signature both scientifically puzzling and scientifically revealing. Recent research published in Scientific Reports (2025) has further complicated the picture, showing that Alzheimer''s itself can be divided into at least three pathological subtypes with four or more clusters identifiable by tau PET and MRI, and concordance between biological staging and clinical staging running at only 30 to 50%. If different biological entities are being grouped under a single treatment umbrella, the repeated failure of single-target Alzheimer''s drugs becomes structurally predictable — just as it would be scientifically incoherent to treat all lung cancers with the same agent regardless of molecular driver. This is precisely the paradigm shift oncology already completed, and dementia research may be approaching its equivalent inflection point.
Dementia''s Coming Demographic and Economic Crisis
The global scale of dementia transforms the karyoptosis discovery from an interesting scientific finding into a matter of urgent public health and economic significance. In 2019, 57.4 million people worldwide were living with dementia; by 2050, that figure is projected to reach 152.8 million — a 2.7-fold increase — with the sharpest growth anticipated in North Africa and the Middle East (367% increase) and Eastern sub-Saharan Africa (357% increase). The cumulative global macroeconomic burden of Alzheimer''s disease from 2020 through 2050 is estimated at $14.513 trillion in constant 2020 dollars, equivalent to 0.421% of annual global GDP, with the largest national burdens falling on China ($2.96 trillion), the United States ($2.33 trillion), and Japan ($1.76 trillion). Informal caregiving — unpaid labor by families — accounts for 60 to 85% of total dementia costs depending on country income level, with per-person economic costs reaching $7,514 annually in high-income nations. Lecanemab, the current gold standard of approved disease-modifying therapy, provides roughly one year of slowed progression over a four-year treatment window — while introducing a brain swelling risk of 119 per 1,000 treated patients versus 12 per 1,000 on placebo. A therapy capable of delaying dementia onset by even five years across a meaningful share of the patient population would produce trillion-dollar downstream economic savings, primarily through reduced informal caregiving costs — making karyoptosis research not just a scientific priority but an economic imperative.
Positive & Negative Analysis
Positive Aspects
- Illuminating a Blind Spot in 40 Years of Brain Research
The most significant contribution of the karyoptosis discovery is that it proves a major pathway of neuronal death has existed, unrecognized, throughout the entire era of modern Alzheimer''s research. For decades, the scientific community operated under the assumption that neuronal loss in Alzheimer''s was adequately explained by amyloid-related toxicity and established death pathways like apoptosis — but the observation that karyoptosis signatures appear in 35% of frontal cortex neurons in Alzheimer''s patients directly challenges that assumption in a way that cannot be easily dismissed. If a third of the relevant neurons are dying through a mechanism we did not know existed, then decades of drug development efforts were operating on an incomplete map of the disease — and the 99.6% clinical trial failure rate becomes dramatically less mysterious in retrospect. This discovery also opens the conceptual door to analogous unmapped death pathways in other neurodegenerative diseases: if karyoptosis went undetected in Alzheimer''s research for 40 years, there may be equivalent blind spots in Parkinson''s disease, ALS, and other conditions where large-scale unexplained neuronal loss is a central feature. The scope of what this finding implies for the broader field of neurodegeneration research extends well beyond any single therapy or diagnostic tool developed in its wake.
- Drug Repurposing Could Compress the Clinical Timeline Significantly
One of the most practically significant aspects of the karyoptosis finding is that it points directly toward therapeutic targets — p38 MAP kinase and LaminB1 — for which pharmacological tools already exist in various stages of human clinical development. Neflamapimod, MW150, and VX-745 all have human safety data accumulated through prior clinical trials in Lewy body dementia, Alzheimer''s disease, and mild cognitive impairment, meaning that repurposing these compounds toward karyoptosis-targeted applications could bypass Phase 1 safety studies entirely — potentially saving three to five years of development time. Drug repurposing already represents 33% of the 2025 Alzheimer''s clinical pipeline, confirming this is a validated and widely accepted development strategy rather than a theoretical shortcut. The average time from molecule discovery to approved drug runs 12 to 15 years in conventional development; repurposing established compounds can compress that to seven to ten years in favorable scenarios. For the 57.4 million people currently living with dementia and the families caring for them, that time compression is not an abstract efficiency gain — it is the difference between a treatment arriving in time to help and arriving too late.
- Shared Mechanism Creates a Two-Disease Treatment Opportunity
The fact that karyoptosis was identified in both Alzheimer''s disease and frontotemporal dementia creates an unusually attractive clinical development opportunity: a single therapeutic strategy could potentially address two distinct patient populations through a single mechanism target. FTD currently has no approved disease-modifying therapy whatsoever, meaning the unmet medical need is absolute — and a drug that demonstrates efficacy against karyoptosis in FTD could represent the first ever disease-modifying treatment for a condition affecting 10 to 20% of all dementia patients worldwide. For drug developers, covering two indications with a single compound improves the commercial case for investment and potentially allows clinical trials to be designed with adaptive protocols serving both populations simultaneously. Dr. Fanto framed this as a key element of the discovery, noting that researchers found "a relatively rare disease shares a mechanism with a condition affecting millions" — a formulation that precisely captures the therapeutic leverage this shared pathway creates. As the 2030 Alzheimer''s treatment market is projected at $13.7 billion, an FTD market addressable by the same compound adds meaningfully to the financial investment case — which in turn could accelerate the timeline to actual patient access.
- Foundation for a Much-Earlier Intervention Paradigm
The 35% prevalence of karyoptosis signatures in Alzheimer''s frontal cortex neurons strongly implies that this mechanism is active well before patients present with symptoms severe enough to qualify for current treatment criteria — and that implication carries enormous consequences for how the field thinks about when to intervene. The current standard of care initiates pharmacological treatment only after cognitive decline has been clinically documented, at which point substantial neuronal loss has already occurred through multiple pathways. If karyoptosis is operating from early in the disease trajectory, effective interception would require intervention years before the first clinic visit — a fundamental shift in the logic of dementia medicine. The FDA''s 2025 approval of blood-based p-tau 217 and Abeta42 diagnostic assays has already made earlier biological detection possible; adding a karyoptosis-specific biomarker to that toolkit would enable identification of patients at high risk of karyoptosis-driven neuronal loss while their brains still retain substantial capacity. This represents not just a new drug target but a new philosophy: the shift from treating disease after it has become symptomatic to preventing it from reaching the clinical threshold in the first place — the same logic that transformed cancer outcomes through population-level screening programs applied consistently over decades.
Concerns
- 28 Patients — A Preliminary Evidence Base with Real Limitations
The most straightforward limitation of the karyoptosis discovery is the size of the study''s patient cohort. Analyzing approximately 3,000 cells from the post-mortem brain tissue of 28 patients with Alzheimer''s or frontotemporal dementia is an entirely appropriate scope for establishing a new mechanism''s existence — but it is not a sufficient evidentiary foundation for drug development decisions, treatment standard changes, or confident epidemiological claims about prevalence. To establish karyoptosis as a genuine therapeutic target rather than an interesting biological observation, multi-center replication studies involving hundreds to thousands of patients across diverse racial, genetic, and clinical backgrounds are essential, and such studies take two to three years at minimum to design, fund, recruit, and analyze. The historical record in Alzheimer''s research provides sobering precedent: numerous promising targets identified in small early studies failed to replicate at scale, and the 99.6% failure rate between 2002 and 2012 is partly a record of biomarkers and mechanisms that appeared compelling in limited sample sets before collapsing under the weight of larger, more rigorous investigation. The gap between "discovered in a promising study" and "validated as a therapeutic target" is precisely where many of the past 40 years of Alzheimer''s failures actually occurred, and there is no scientific reason to expect karyoptosis to be exempt from that historical pattern.
- Causality Is Not Established — Correlation in Post-Mortem Tissue Has Limits
The karyoptosis study is, at its core, an observational analysis of post-mortem brain tissue from late-stage patients — which means it can demonstrate that karyoptosis is present in Alzheimer''s and FTD brains in elevated proportions, but cannot definitively establish whether karyoptosis causes or contributes to neurodegeneration, or whether it is a downstream consequence of pathological processes already well advanced by other means. This distinction matters enormously for therapeutic strategy: if karyoptosis is a causal driver operating from early in the disease, blocking it should meaningfully slow progression; if it is a late-stage artifact produced by cells already comprehensively damaged through other mechanisms, blocking it may have no effect on cognitive trajectory. The mouse neuron experiments in which blocking the p38-LaminB1 interaction reduced karyoptosis signatures provide suggestive causal evidence, but the gulf between mouse neuron physiology and human neurodegeneration has swallowed many seemingly robust preclinical findings. This is precisely the same logical trap the amyloid hypothesis fell into — early evidence compelling enough to justify enormous investment, followed by repeated human trial failure once the causality assumption was tested at scale. Establishing robust causal evidence in human tissue will require longitudinal studies that are significantly harder to execute than cross-sectional post-mortem analysis, and that evidence does not yet exist.
- p38 MAPK Inhibitors Have a Complicated Historical Track Record
The enthusiasm surrounding existing p38 MAPK inhibitors as potential karyoptosis-targeting agents needs to be tempered by an honest assessment of that drug class''s historical clinical performance. p38 inhibitors have been investigated across multiple therapeutic areas — rheumatoid arthritis, inflammatory bowel disease, psoriasis, cardiovascular disease — with limited clinical success, and the anti-inflammatory applications that first attracted interest in this target class have not produced approved drugs despite decades of sustained investment. In the Alzheimer''s and neurodegeneration space specifically, Neflamapimod''s decision to pivot its primary focus to Lewy body dementia rather than Alzheimer''s disease is worth noting — it may signal that p38 inhibition in the most common dementia subtype proved less compelling than the initial hypothesis suggested. Furthermore, the karyoptosis application specifically targets the non-catalytic interaction between p38 and LaminB1 rather than p38''s enzymatic activity directly — meaning existing p38 inhibitors may not be optimally designed for this precise molecular objective, and bespoke molecular engineering may be required, adding development time and cost. Blood-brain barrier permeability challenges remain unresolved for this drug class, and the concentration required to achieve meaningful target engagement in human brain tissue may differ substantially from what mouse experiments suggest is sufficient.
- The Structural Inertia of the Alzheimer''s Research Ecosystem
The hardest obstacle karyoptosis faces may not be scientific but institutional: the Alzheimer''s research ecosystem has enormous built-in momentum toward established targets, and redirecting that momentum requires more than a single compelling paper in a peer-reviewed journal. The current pipeline includes 182 active clinical trials and 138 candidate compounds, the majority positioned around amyloid, tau, or neuroinflammatory targets that have attracted billions in development investment and represent years of academic careers, institutional commitments, and commercial strategies built around existing frameworks. Lecanemab''s developer and donanemab''s developer have no rational financial incentive to redirect resources away from therapies projected to generate $3.5 billion and $2 billion respectively in 2030 revenues by adopting an unproven alternative mechanism. Academic resistance can be equally stubborn: the scientific community''s response to the Cochrane Review''s negative verdict on amyloid therapy included significant pushback from researchers whose professional identities are inseparable from the amyloid hypothesis, and a similar dynamic is likely to emerge around karyoptosis. The history of paradigm shifts in medicine is instructive — they rarely occur simply because new data arrives; they typically require the gradual generational transition as the scientific cohort built on the old paradigm yields to researchers trained under different assumptions. That process takes time that dementia patients today simply do not have.
Outlook
The next six months will be dominated by an academic validation battle, and it will not be gentle. The Nature Communications paper is barely two weeks old at time of writing, and thousands of researchers whose careers are built on amyloid hypothesis-based work are not going to embrace this result without pushing back. When the 2026 Cochrane Review concluded that amyloid clearance produces no meaningful clinical benefit, a significant segment of the scientific community immediately pushed back — arguing the meta-analysis unfairly combined effective and ineffective drugs in the same analysis. Expect a similar pattern of criticism directed at karyoptosis: "Twenty-eight patients is too small a sample," "this is a late-stage artifact, not a causal driver," "mouse neuron data does not translate to human pathology" are all scientifically legitimate objections that deserve rigorous answers. I expect at least two or three independent research groups to begin replication attempts in their own patient cohorts within three to six months. Given that this work was funded by the UK Dementia Research Institute, other British dementia research institutions are likely to move fastest, and a preprint or short communication reporting preliminary replication data could land within the year.
The pharmaceutical industry is simultaneously calibrating its response with characteristic speed. The Alzheimer''s treatment market is projected to grow from $2.2 billion in 2020 to $13.7 billion in 2030 — an annual growth rate of roughly 20% — with lecanemab (projected 2030 revenues near $3.5 billion) and donanemab (roughly $2 billion) leading that growth. Both drugs remain fundamentally constrained to amyloid clearance logic, however. If karyoptosis gets validated by independent research groups, it represents the first credible alternative mechanism to receive this level of scientific scrutiny in the Alzheimer''s space, and biotech investors will notice immediately. Among p38 MAPK inhibitors, Neflamapimod is the most clinically advanced, currently running Phase 2 for Lewy body dementia across 160 patients at 29 centers. An attempt to expand that indication to Alzheimer''s — with karyoptosis evidence as scientific rationale — could realistically surface within months of successful replication. My assessment is that at least one biotech company will formally announce a karyoptosis preclinical program within the next six months, even ahead of independent replication results being published.
The six-month to two-year window is where real scientific momentum either builds or collapses entirely. For meaningful clinical progress, karyoptosis must first be replicated not in 28 patients but in multi-site cohorts of hundreds, drawn from diverse racial and genetic backgrounds with varying clinical profiles. A decisive research question is whether karyoptosis activity differs significantly across Alzheimer''s pathological subtypes — typical Alzheimer''s, hippocampal-sparing, and limbic-predominant variants. If karyoptosis proves subtype-specific, that finding becomes the scientific foundation for precision medicine in dementia: stratifying patients by karyoptosis biomarker status and matching each group to a targeted therapeutic strategy. The FDA''s 2025 approval of blood-based p-tau 217 and Abeta42 diagnostic tests already moved early detection one meaningful step forward; integrating a karyoptosis-specific biomarker into that diagnostic framework could substantially improve patient risk stratification accuracy. The 182 active Alzheimer''s clinical trials representing a combined 50,109 enrolled participants are an enormous infrastructure investment — and the emergence of a validated new pathway will inevitably influence how that infrastructure gets reallocated across the research system.
The most realistic near-term clinical scenario, in my view, is the rise of combination therapy as a guiding framework. The amyloid hypothesis is not wrong — it is incomplete. If karyoptosis and amyloid toxicity both contribute to neuronal death through different mechanisms, the next logical step is combining existing anti-amyloid agents with karyoptosis pathway inhibitors rather than treating them as competing approaches. The 2025 Alzheimer''s pipeline already reflects the field''s growing openness to multi-target strategies: more than half of the 138 candidate compounds target non-amyloid mechanisms, including neuroinflammation and immunity (17%), tau-related pathways (11%), and metabolic processes (6%). Adding a p38 MAPK-LaminB1 karyoptosis axis to that mix could open the door to an Alzheimer''s "cocktail therapy" model — directly analogous to how HIV management was transformed from a death sentence into a chronic manageable condition through combination antiretroviral regimens targeting the virus at multiple points simultaneously. That comparison is not hyperbolic; it reflects a genuine structural parallel between where dementia treatment stands today and where HIV treatment stood in the early 1990s. For this scenario to be testable within two years, Phase 1 safety data for a karyoptosis-targeted compound must emerge, and repurposing existing p38 inhibitors with established human safety profiles represents the fastest credible route.
Looking two to five years out, the deepest potential impact of this discovery may be a fundamental reclassification of what we actually call Alzheimer''s disease. Today, 57.4 million people worldwide live with dementia, and that figure is projected to reach 152.8 million by 2050 — a 2.7-fold increase. Behind those numbers is a pathologically heterogeneous collection of conditions currently grouped under diagnostic labels that may not reflect underlying biological reality. If karyoptosis proves subtype-specific within Alzheimer''s, the design of future clinical trials will require substantial changes: rather than enrolling "Alzheimer''s patients" as a uniform population, researchers will need to separate karyoptosis-positive and karyoptosis-negative cohorts and apply different interventions to each. This mirrors the paradigm shift oncology already completed — where lung cancer is no longer treated as a single monolithic disease but stratified by EGFR mutation, ALK rearrangement, KRAS status, and more, with each molecular subtype matched to its most effective targeted therapy. Dementia research may be approaching an equivalent inflection point. The urgency is particularly acute in regions projected to see the sharpest increases in dementia burden: North Africa and the Middle East at a projected 367% increase, and Eastern sub-Saharan Africa at 357% — both regions where diagnostic infrastructure is often minimal, making scalable biomarker-based early detection technologies a geopolitical and humanitarian priority, not merely a scientific one.
The economic stakes are substantial enough to transform this from a scientific story into a public policy priority. The cumulative global macroeconomic burden of Alzheimer''s disease from 2020 through 2050 is estimated at $14.513 trillion in constant 2020 dollars — equivalent to 0.421% of annual global GDP. China carries the largest projected burden at $2.96 trillion, followed by the United States at $2.33 trillion and Japan at $1.76 trillion. Informal caregiving — unpaid labor provided by families — accounts for 60 to 85% of total dementia costs depending on a nation''s income level, meaning the financial impact is distributed far beyond hospital systems into individual households globally. If karyoptosis-targeted early intervention could delay symptom onset by even five years across a meaningful share of the patient population, the downstream economic savings would reach into the hundreds of billions — potentially much more. That remains speculative, but the possibility is now real in a way it was not six months ago.
To put a probability framework on what comes next: the optimistic scenario has independent replication succeeding within 12 to 18 months, a repurposed p38 inhibitor entering Phase 2 for karyoptosis-targeted Alzheimer''s therapy within three years, and a karyoptosis-specific biomarker getting integrated into existing clinical diagnostic panels — together opening a genuine early-intervention window that current approved drugs simply cannot access. The base scenario has replication eventually succeeding but clinical translation taking five to seven years, with karyoptosis inhibition ultimately becoming a component of combination therapy alongside anti-amyloid agents, pushing the effective treatment benefit from the current one-year delay ceiling toward two or three years. The pessimistic scenario has replication failing — or establishing karyoptosis as a terminal-stage artifact rather than a causal driver — with p38 inhibition ultimately failing to demonstrate meaningful benefit in human brain tissue, adding this discovery to the long and painful list of promising preclinical leads that could not survive the translation gap. I weight these scenarios at roughly 20% optimistic, 50% base, and 30% pessimistic. The decisive variable is the two-year replication outcome — if that fails, the rest becomes moot. For anyone reading this with a family history of dementia: the blood-based diagnostic tests already approved in 2025 are available now, today. Talk with your physician. Karyoptosis is a potential future. Early detection is a lever you can actually pull right now.
Sources / References
- Karyoptosis mediates cell death and neurodegeneration upon proteotoxic stress — Nature Communications
- New mechanism found for neuronal death in Alzheimer''s and FTD — King''s College London
- New mechanism found for neuronal death in Alzheimer''s and FTD — EurekAlert! / AAAS
- Anti-amyloid monoclonal antibodies for Alzheimer''s disease — Cochrane Library
- Scientists discover mechanism for brain cell death in Alzheimer''s disease — Alzheimer''s Research UK
- Anti-amyloid monoclonal antibodies: systematic review of efficacy and safety — PubMed Central
- The global macroeconomic burden of Alzheimer's disease — Lancet Global Health
- Alzheimer''s disease drug development pipeline: 2025 — PMC / Alzheimer''s & Dementia